导语

急性心肌梗死(AMI)是世界范围内发病率和死亡率的主要原因。及时冠脉血液再灌注是限制心肌梗死、维持心功能的最有效治疗方法。然而,再灌注往往会加重心脏损伤,这种现象被称为缺血再灌注(I/R)损伤。尽管已经对其潜在的分子机制进行了大量的研究,但临床上减轻心脏I/R损伤的可行治疗靶点仍然缺乏。云序生物acRIP-seq助力哈尔滨医科大学杨宝峰院士团队在Redox Biology期刊(IF=10.7)发表题为“The positive feedback loop of the NAT10/Mybbp1a/p53 axis promotes cardiomyocyte ferroptosis to exacerbate cardiac I/R injury”的研究论文。这是继上期该队团在Advanced science期刊(IF=14.3)发表题为“The m7G Methyltransferase Mettl1 Drives Cardiac Hypertrophy by Regulating SRSF9-Mediated Splicing of NFATc4”的m7G修饰研究后,该课题组又一个RNA修饰(ac4c)研究。在本研究中,作者发现I/R诱导的NAT10上调是由p53以转录依赖的方式调控的。使用功能获得和功能丧失策略,作者确定了NAT10的促铁效应是心脏I/R损伤的主要触发因素。NAT10通过引起Mybbp1a mRNA的ac4C修饰增强了其稳定性,从而导致p53的激活,进而抑制抗铁沉基因SLC7A11的转录。这些发现表明,靶向NAT10/Mybbp1a/p53轴是治疗心脏I/R损伤的一个有希望的新方向。本次研究中,云序生物有幸提供了ac4C acRIP-seq,RNA-seq以及生信分析。

研究摘要
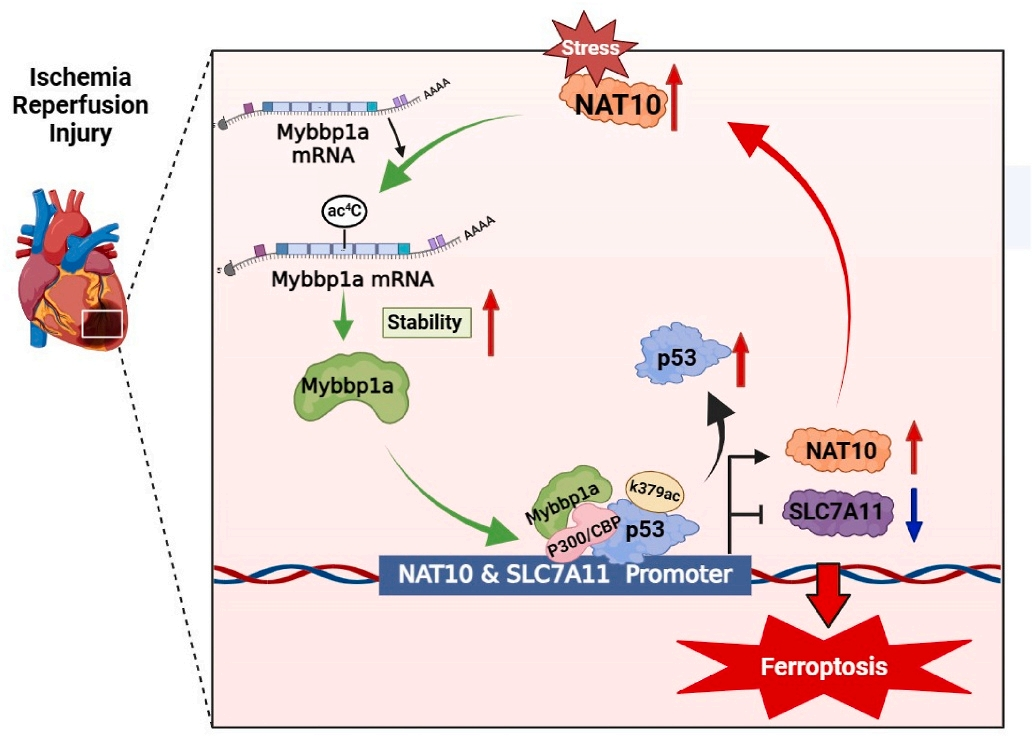
技术路线

研究结果

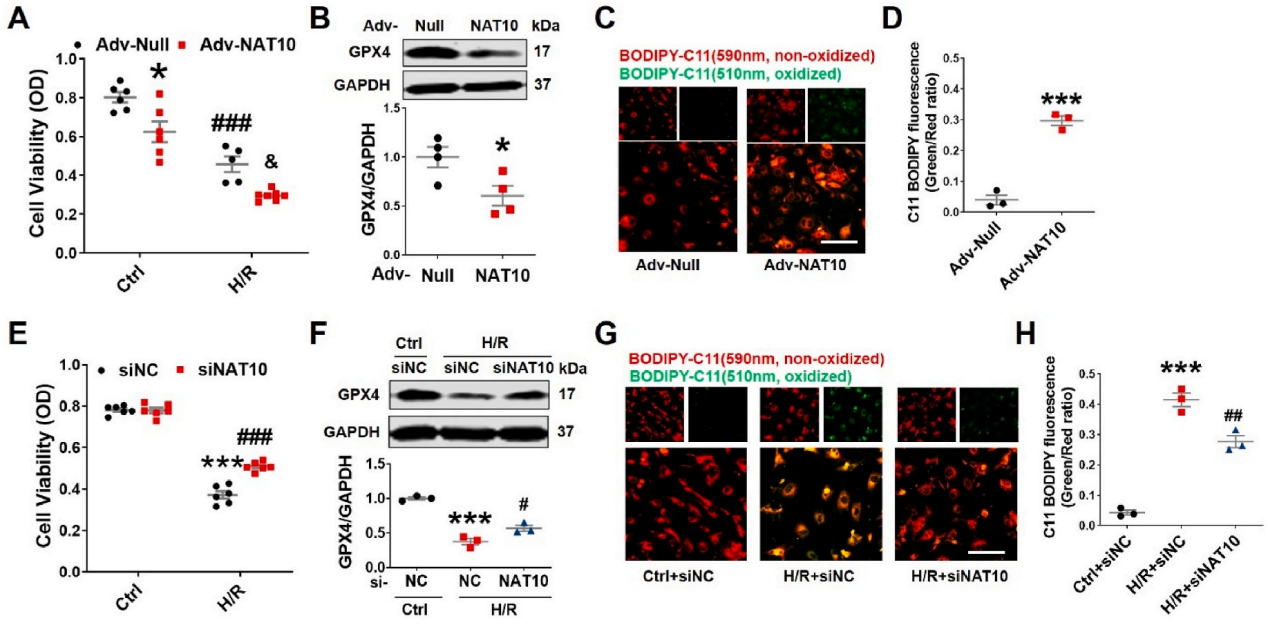
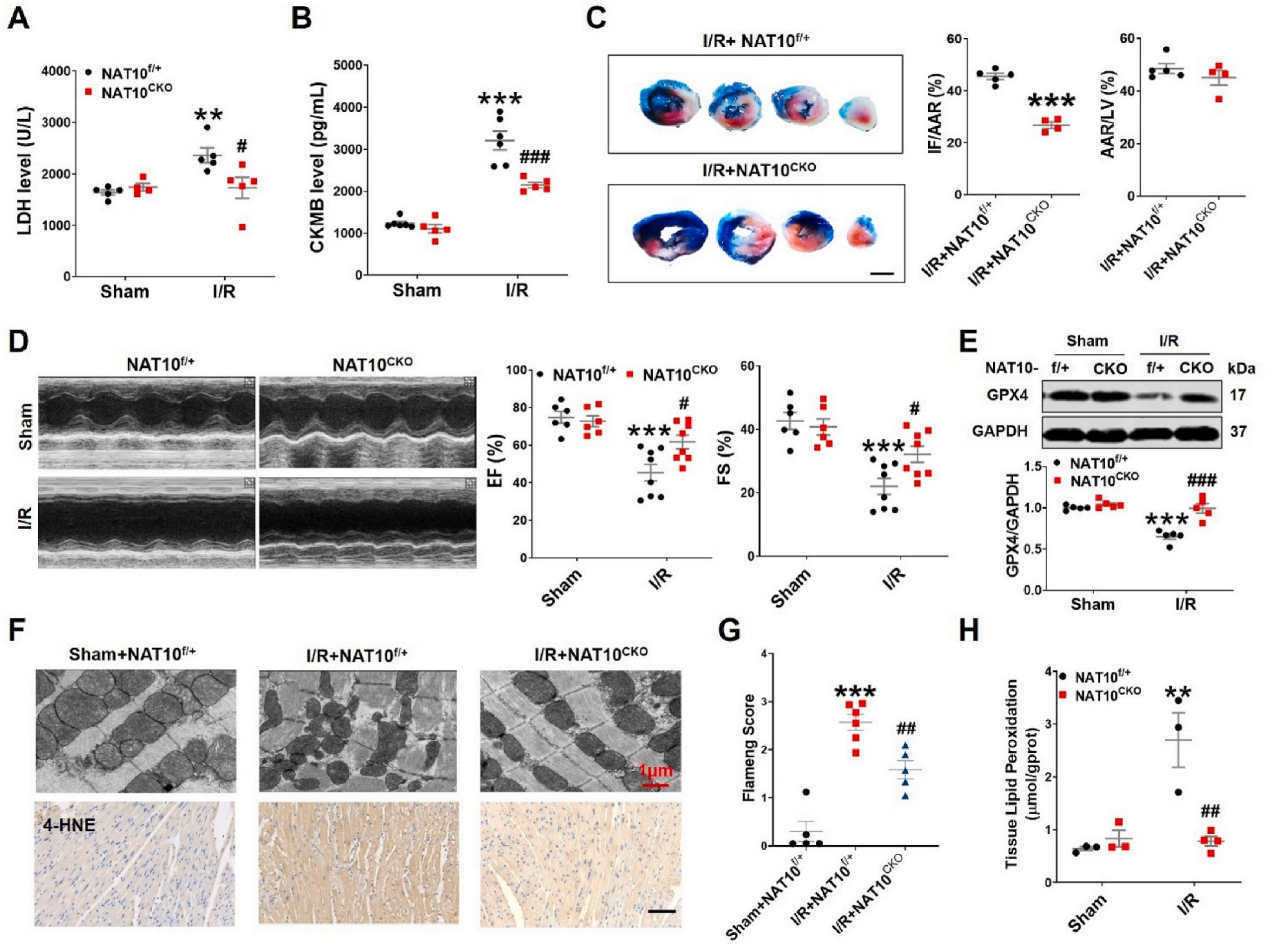
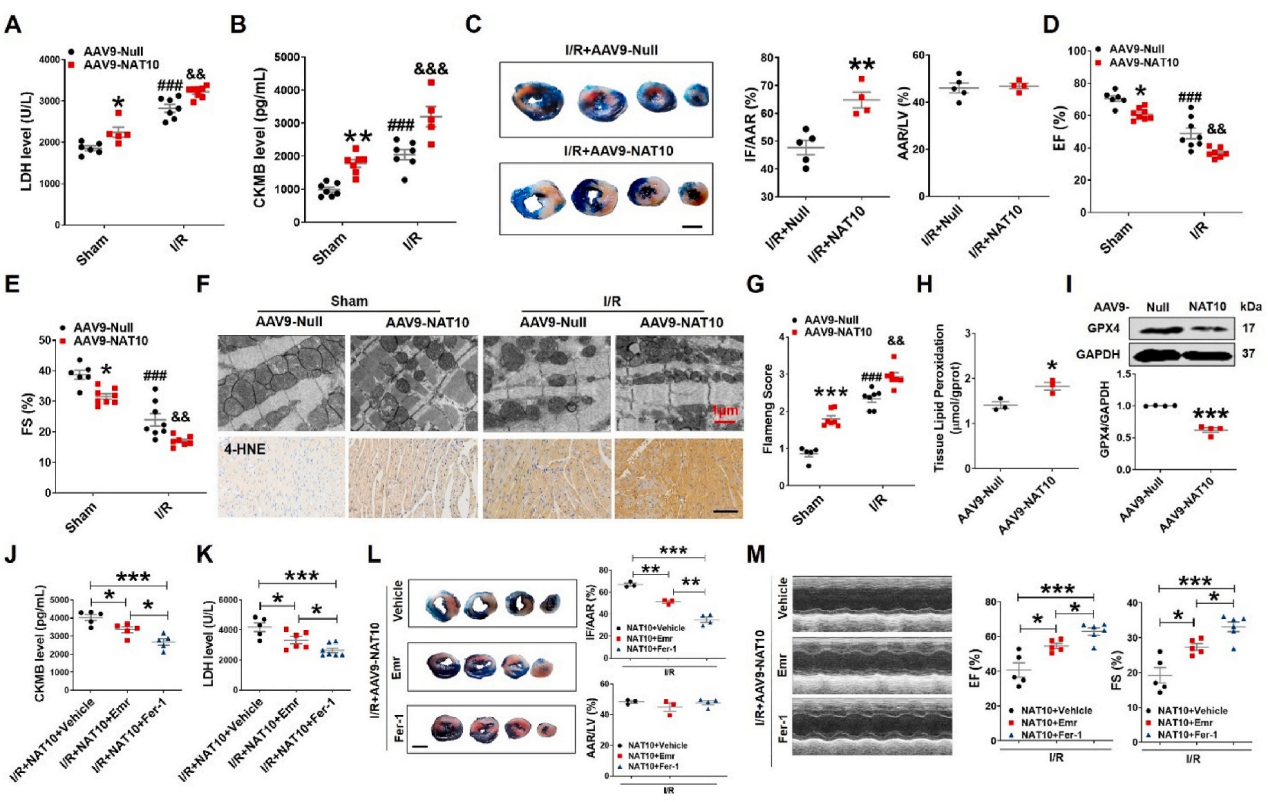
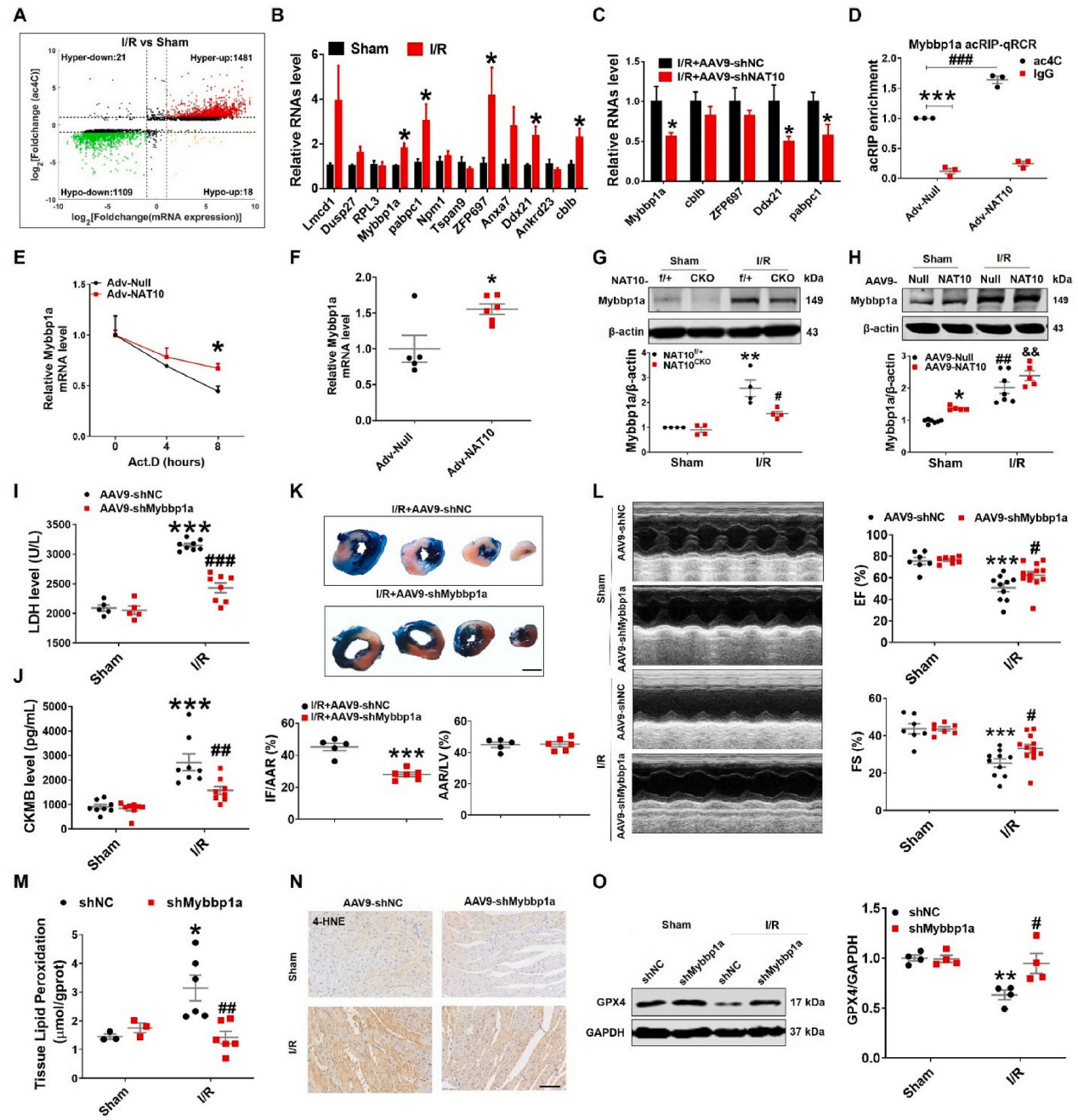
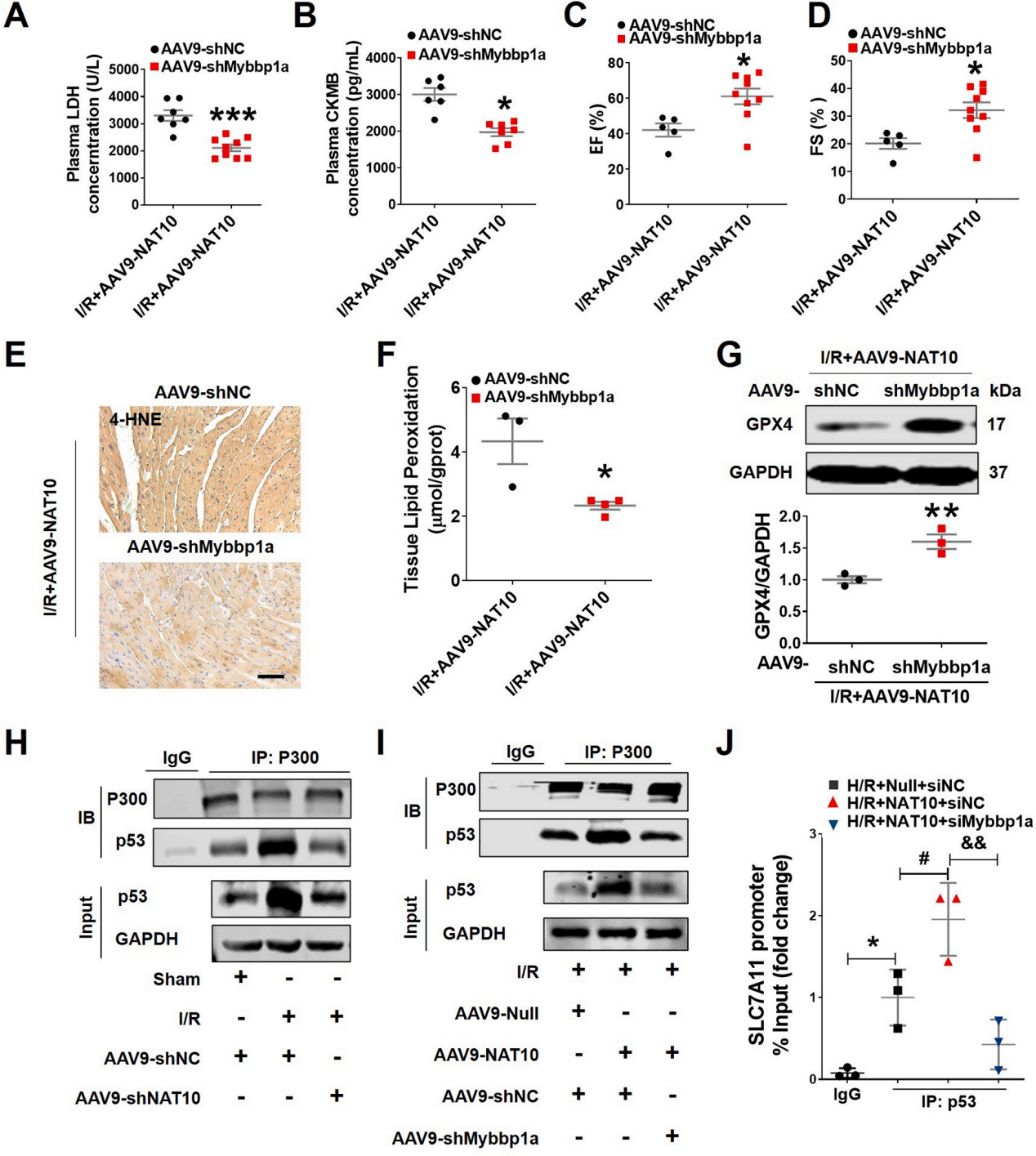
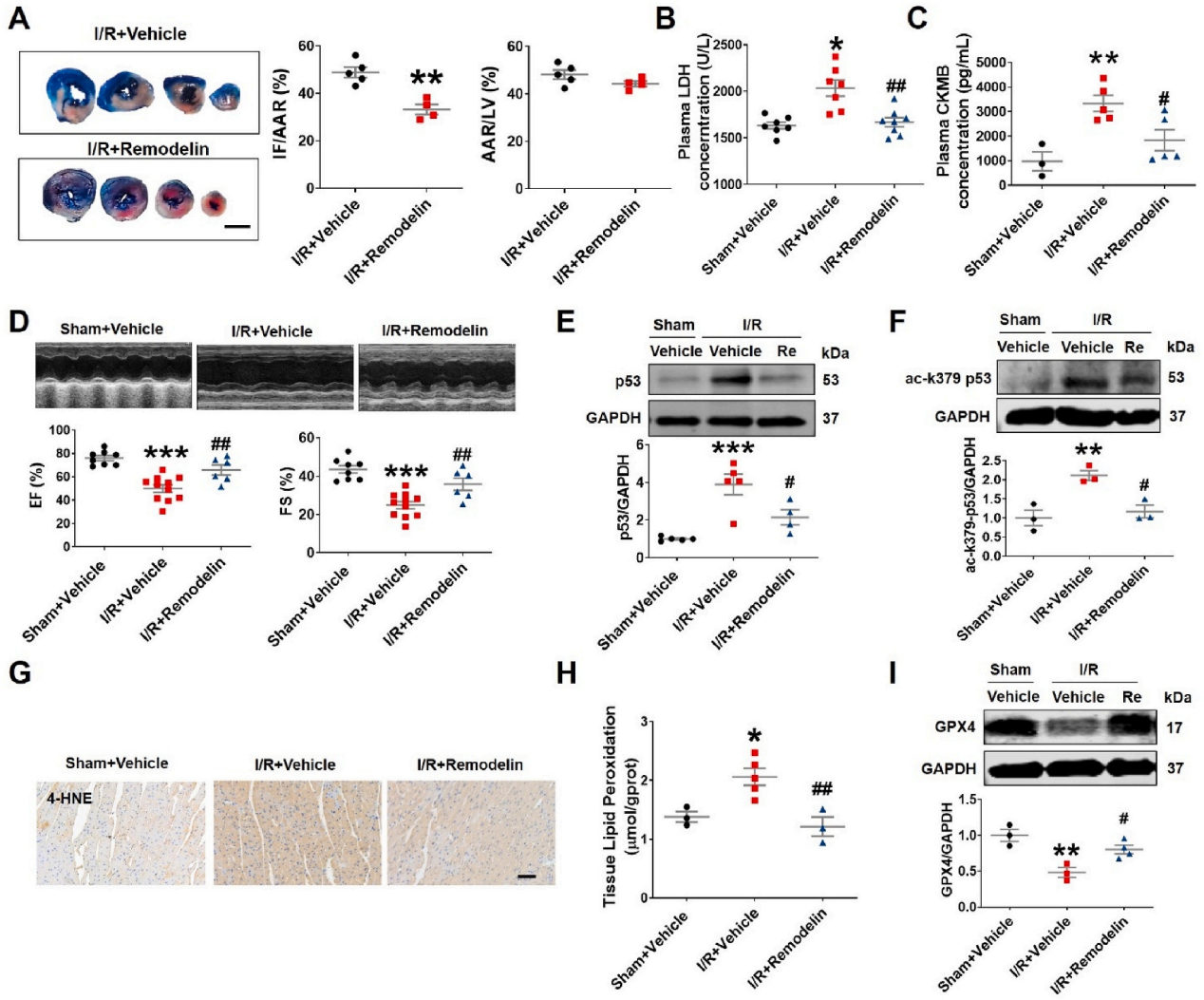
全文小节
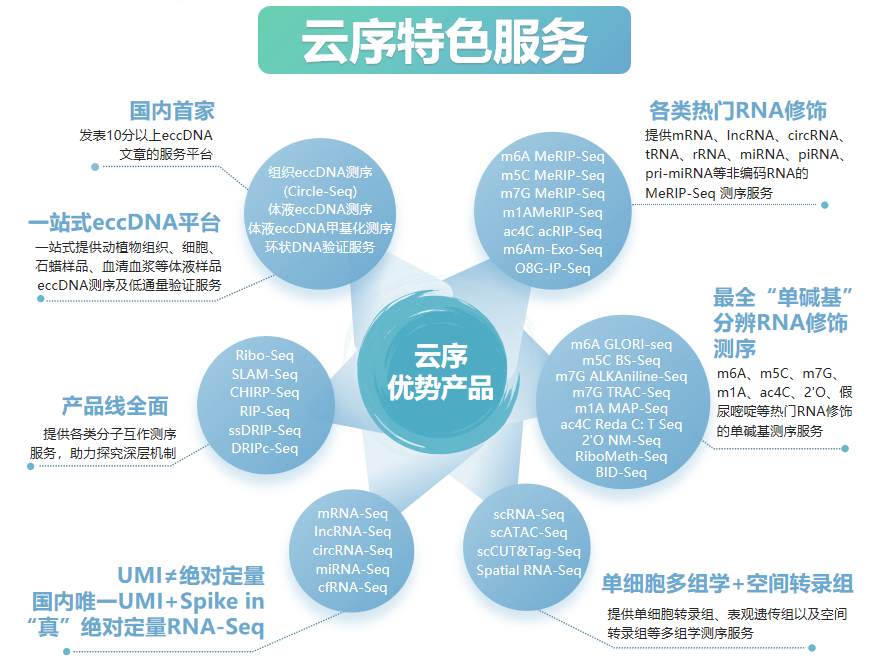
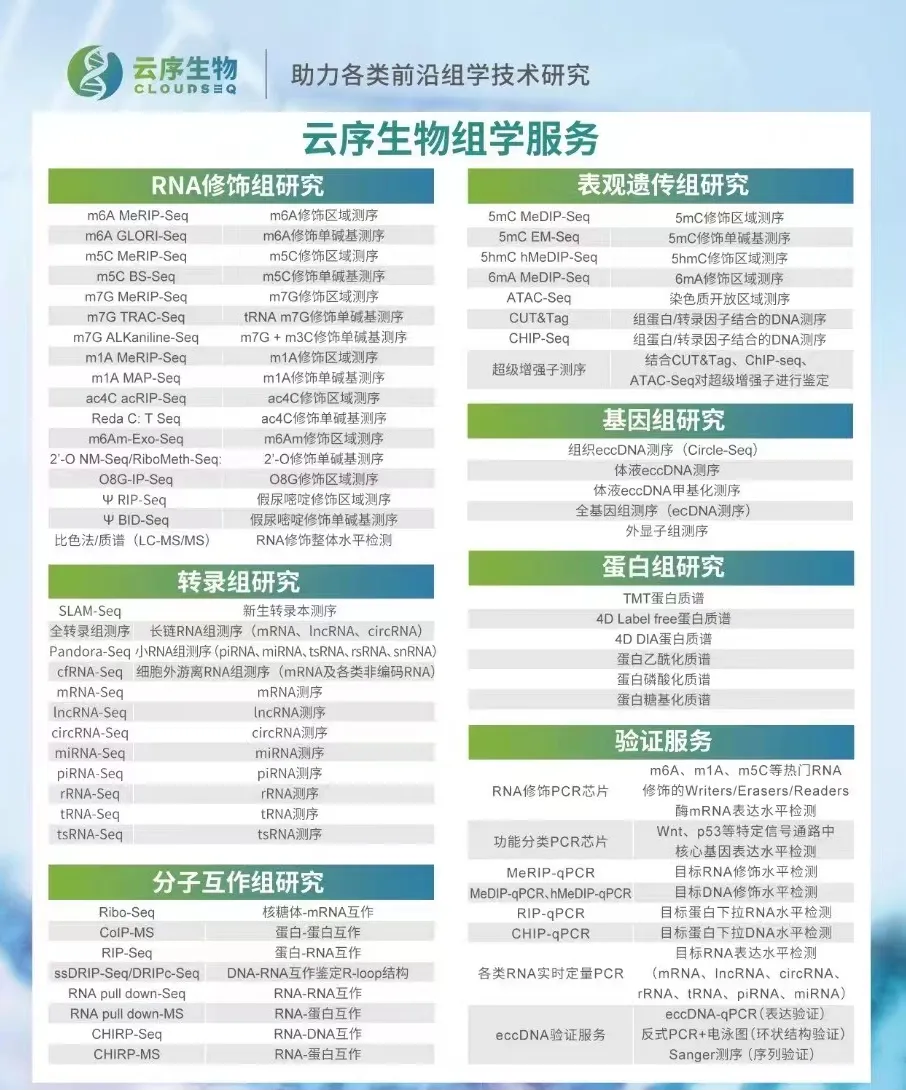
云序生物RNA修饰优势
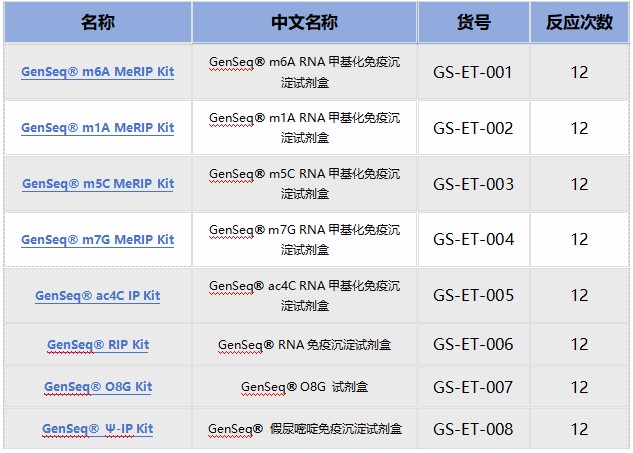
优势一:RNA修饰产品线最全,热门修饰IP类/单碱基分辨测序 + IP试剂盒
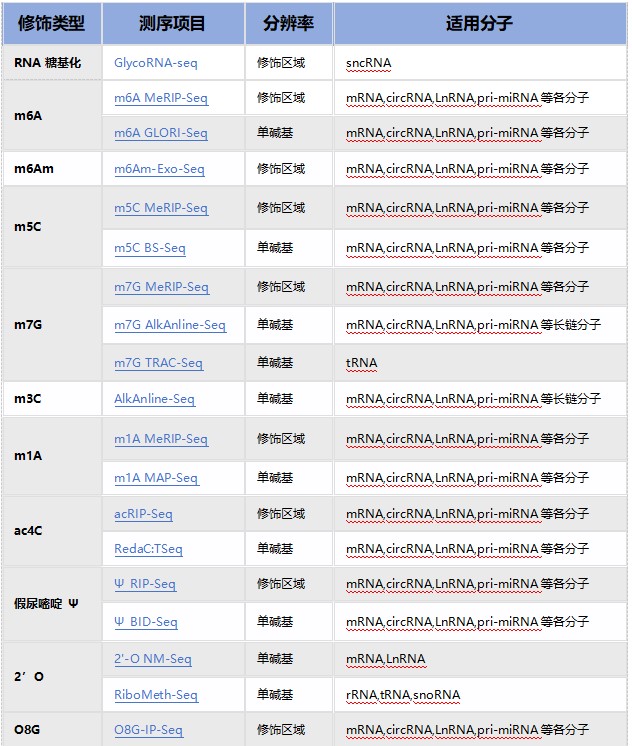
优势二:数千篇SCI论文、数万例测序样本
优势三:MeRIP-seq全面升级,spike in严格质控IP实验

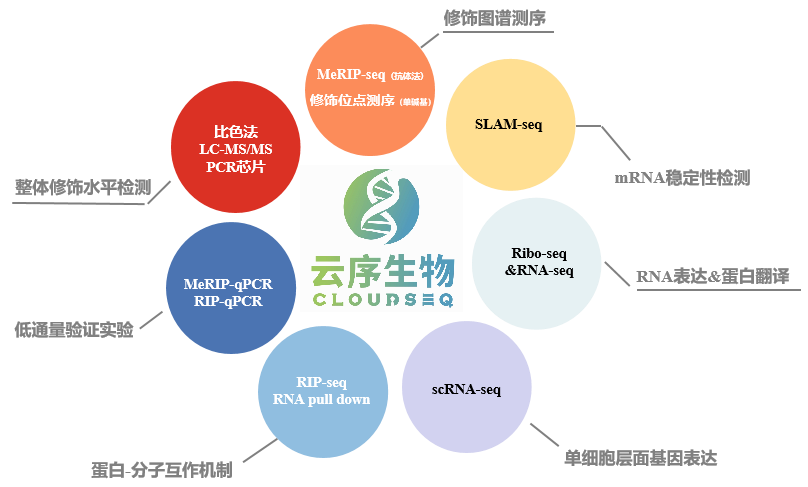
优势四:测序到验证、靶标定位到分子机制,云序提供一站式服务
往期回顾
黄荷凤院士团队前沿成果 | 云序m6A MeRIP-Seq&RIP-seq技术助力揭示高龄父亲引起子代神经炎症风险加剧
聚焦表观,转录共荣!墨卓生物与云序生物达成合作
PNAS/Q1 云序生物助力苏大陶金团队揭示m6A介导神经病理性疼痛新机制
IF=20.4 高分ac4C新鲜出炉||云序助力客户探究ac4C与肿瘤和免疫相关新机制
Get新热点! m6A&单细胞强强联合,助力文章升级! | m6A&单细胞专题一
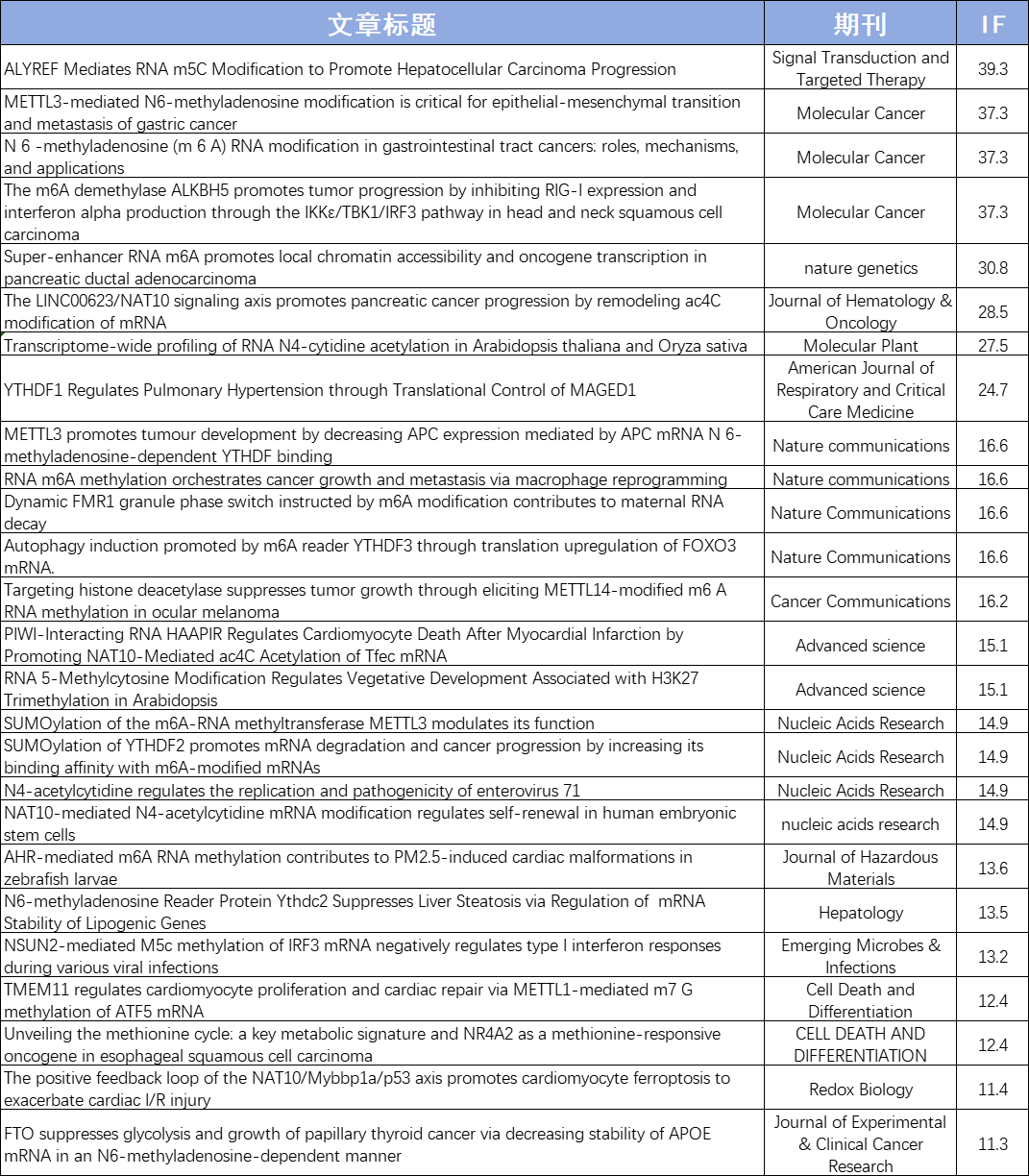
云序客户近期RNA修饰高分文章速览
Nature Genetics IF=30.8|非编码seRNA m6A修饰介导组蛋白修饰和癌基因表达
云序客户| m6A MeRIP-seq助力揭示早发糖尿病表观调控新机制
云序客户m6A高分文章|揭示组蛋白乙酰化与m6A修饰在眼部黑色素瘤发生中的共同作用机制

下期精彩继续